U.S. Food and Drug Administration (FDA)
The FDA does not follow the NIH Page Limitation Guidelines or the NIH Review Criteria. Applicants are encouraged to consult with FDA Agency Contacts for additional information regarding page limits and the FDA Objective Review Process.
Center for Drug Evaluation and Research (CDER)
Developing Biomarkers for Trastuzumab-induced Cardiotoxicity
R30 Preventive Health Service Venereal Disease Research, Demonstration, and Public Information and Education Grants
- June 7, 2017 - Notice of Change to Key Dates for RFA-FD-17-006. See Notice NOT-FD-17-011.
RFA-FD-17-006
none
93.103
This award is intended to provide support to conduct clinical studies at a clinical site with capability and patients samples to investigate potential biomarker (cMLC1) for trastuzumab-induced cardiotoxicity.
1) There will be an establishment of the assay to quantitatively measure cMLC-1 in humans.
2) Investigations will be conducted to determine the value of cMLC-1 in humans as an early predictor of cardiotoxicity in a pilot study. A comparison will be done of the level of cMLC-1 in 5 patients who subsequently developed cardiotoxicity and 5 patients who did not. Patients will be matched for age. Cardiotoxicity was defined using the Cardiac Review and Evaluation Committee for Trastuzumab (CREC) criteria as a decrease of more than 10% in the echocardiographic left ventricular ejection fraction to a value of less than 55%.
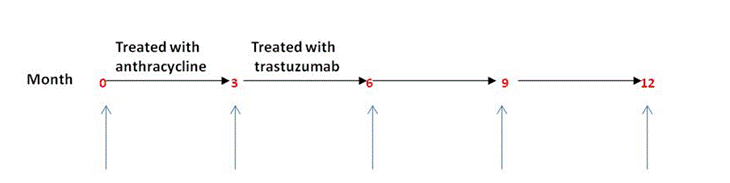
3) Women were monitored every 3 months for 15 months.
4) The archived plasma samples collected at multiple time points indicated below schematically will be tested for the biomarker cMLC-1 and for troponin I, an established marker for heart muscle damage as a control.
May 22, 2017
May 22, 2017
Not Applicable
New Dates July 24, 2017, by 11:59 PM Eastern Time.
RFAs that must be reviewed on a compressed timeline and want to prohibit late applications must add the following text prior to approval for publication, here and below.
No late applications will be accepted for this Funding Opportunity Announcement.
Applicants are encouraged to apply early to allow adequate time to make any corrections to errors found in the application during the submission process by the due date.
Applicants should be aware that on-time submission means that an application is submitted error free (of both Grants.gov and eRA Commons errors) by 11:59 PM Eastern Time on the application due date.
Late applications will not be accepted for this FOA.
Not Applicable
July, 2017
Not Applicable
September 1, 2017
July 11, 2017
Not Applicable
It is critical that applicants follow the instructions in the SF424 (R&R) Application Guide, except where instructed to do otherwise (in this FOA or in a Notice from the NIH Guide for Grants and Contracts). Conformance to all requirements (both in the Application Guide and the FOA) is required and strictly enforced. Applicants must read and follow all application instructions in the Application Guide as well as any program-specific instructions noted in Section IV. When the program-specific instructions deviate from those in the Application Guide, follow the program-specific instructions. Applications that do not comply with these instructions may be delayed or not accepted for review.
Part 1. Overview Information
Part 2. Full Text of the Announcement
Section
I. Funding Opportunity Description
Section II. Award Information
Section III. Eligibility Information
Section IV. Application and Submission
Information
Section V. Application Review Information
Section VI. Award Administration Information
Section VII. Agency Contacts
Section VIII. Other Information
Breast cancer is one of the most common cancers among women in the United States. It is the second leading cause of cancer death in women, after lung cancer. Trastuzumab (also known as Herceptin ) is a humanized monoclonal antibody against extracellular domain IV of the human epidermal growth factor receptor 2 (HER2) protein and is approved for the treatment of breast cancers that are HER2 positive. Trastuzumab provides considerable therapeutic benefits in HER2 positive breast cancers and improves disease free and overall survival after adjuvant chemotherapy. However, trastuzumab treatment is also associated with cardiac dysfunction (1), mechanisms of which are unclear. The incidence of cardiotoxicity is increased when trastuzumab is used in combination with anthracyclines, such as doxorubicin, which are a class of chemotherapeutic agents with demonstrated efficacy in the breast cancer setting (2, 3). Therefore, a general consensus has emerged that the concurrent use of trastuzumab with an anthracycline should be avoided (4). Given the considerable antitumor effect of trastuzumab in HER-2-positive breast cancer and the importance of combining trastuzumab with an anthracycline for optimal oncologic efficacy, it is important to understand the mechanisms contributing to cardiac toxicity by these drugs and to identify potential biomarkers to help define the risks and benefits of trastuzumab treatment in the adjuvant treatment of breast cancer, as well as to provide preventive treatment to reduce the incidence of cardiac toxicity.
Trastuzumab treatment is associated with cardiac dysfunction, which manifests as the decrease in the left ventricular ejection fraction (LVEF) and heart failure (HF) (1, 5, and 6). Trastuzumab-induced cardiotoxicity was reported to occur in up to 7% of patients when trastuzumab is used as a single agent (7). When combined with an anthracycline, cardiotoxicity is notably increased and has been reported to occur in up to 27% of patients (7). Current clinical management for trastuzumab-induced cardiotoxicity relies on the use of echocardiography to detect the reduction in LVEF. Based on the extent of LVEF reduction, a decision regarding continuation or discontinuation of trastuzumab therapy is made. However, reduction in LVEF is a part of the late phase of left ventricular dysfunction, which occurs as a compensatory mechanism of the heart to preserve contractility. There are no clinically approved biomarkers that can be used to predict the cardiac dysfunction induced by trastuzumab. Troponin 1 is considered as a sensitive or specific biomarker in the diagnosis of myocardial infarction. However, it is not sensitive and specific for the diagnosis of trastuzumab-induced cardiotoxicity. Using echocardiography, we found that trastuzumab significantly reduced LVEF in mice. Importantly, this reduced LVEF in mice was associated with elevated levels of troponin 1 and cardiac myosin light chain 1 (cMLC-1) in mice sera compared to troponin 1 and cMLC-1 levels in control mice. cMLC-1 has been implicated as a marker of heart function and may be a promising laboratory marker with diagnostic potential for the assessment of minimal myocardial damage (8). Early identification of patients who are at risk for left ventricular dysfunction following trastuzumab therapy could lead to a better surveillance strategy and early initiation of potentially cardio protective therapy. Using primary cardiomyocytes, Dr. Wen Jin Wu's lab at FDA recently found that trastuzumab inhibits autophagy of cardiomyocytes and increases the production of reactive oxygen species (ROS) in cardiomyocytes. These data could be potentially used to develop a bioassay for HER2 targeted therapeutics, including monoclonal antibodies, bispecific antibodies, and trastuzumab biosimilars for adverse events.
HER2 is a member of the ErbB/HER family of receptor tyrosine kinases. The ErbB/HER family receptors are involved in the development of numerous human cancers, and have been intensely pursued as therapeutic targets for cancer therapy (9, 10). Trastuzumab is a humanized therapeutic monoclonal antibody directed against the extracellular domain of HER2. Trastuzumab is clinically efficacious either as a single agent or in combination with standard chemotherapy regimens such as anthracyclines. Both anthracyclines and trastuzumab are associated with considerable cardiotoxicity. It has been an open question in the HER2-targeted therapy as to why combining trastuzumab with an anthracycline, the cardiotoxicity is significantly increased. cMLC-1 is part of the myosin complex and plays an important role in cardiac muscle contraction. Impaired integrity of cardiomyocytes leads to release of cMLC-1 from the myocardium into circulation (8, 11, and 12). It has not been reported if the release of cMLC-1 from the myocardium into circulation is clinically related to trastuzumab-induced cardiotoxicity.
Reference
(1) Telli ML, Wittels RM. Trastuzumab-Related Cardiac Dysfunction. Journal of the National Comprehensive Cancer Network 2011;9:243-249.
(2) van Hasselt JG, Boukhout AH, Beijnen JH, et al. Population Pharmacokinetic-Pharmacodynamic Analysis of Trastuzumab-Associated Cardiotoxicity. Nature 2011;90:126-132.
(3) Slamon DJ. Leyland-Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 2001;11:783-792.
(4) Ewer MS, Vooletich MT, Durand JB, et al. Reversibility of Trastuzumab-Related Cardiotoxicity: New Insights Based on Clinical Course and Response to Medical Treatment. J Clin Oncol 2005;23:7820-7826
(5) Bowles EJ, Wellman R, Feigelson HS, et al. Risk of heart failure in breast cancer patients after anthracycline and trastuzumab treatment: A retrospective cohort study J Natl Cancer Inst 2012;104:1293 1305.
(6) DeCara DJ. Early detection of chemotherapy-related left ventricular dysfunction. Curr Cardiol Rep 2012;14:334-341
(7) Seidman A. Hudis C, Pierri MK et al. Cardiac dysfunction in the trastuzumab clinical trials experience. J Clin Oncol 2002;20:1215-1221.
(8) ElZarrad MK, Mukhopadhyay P, Mohan N, et al (2013) Trastuzumab alters the expression of genes essential for cardiac function and induces ultrastructural changes of cardiomyocytes in mice. PLoS ONE 2013 8(11): e79543.
(9) Hirsch DS, Wu WJ. Cdc42: an effector and regulator of ErbB1 as a strategic target in breast cancer therapy. Expert Rev Anticancer Ther 2007;7:147-57.
(10) Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer 2005;5:341-54.
(11) Stejskal D, Lacnak B, Andelova K, et al. MLC-1 (myosin light chains-1) in differential diagnosis of dyspnea Biomed Papers 2005;149: 89 91.
(12) Hansen MS, Stanton EB, Gawad Y, et al. Relation of circulating cardiac myosin light chain 1 isoform in stable severe congestive heart failure to survival and treatment with flosequinan. Am Journal of Cardiology 2002;90:969 973.
Regulatory Impact
Office of Biotechnology Product (OBP) is responsible for the product quality review of therapeutic monoclonal antibodies, antibody related product such as antibody-drug conjugates, bispecific antibodies, and biosimilars. Dr. Wu, an Investigator of this proposed study at FDA, is the product quality reviewer responsible for at least six therapeutic monoclonal antibodies and trastuzumab biosimilars targeting HER family receptors, which are currently at different developmental stages. Monoclonal antibodies have become the most successful molecular modality in the development of modern biological medicine in the past decade. The technologies used to generate therapeutic monoclonal antibodies are getting more complicated. In order to fully understand the products and to make right decisions for the products at the different clinical developmental stages, FDA reviewers should be armed with current understanding with regards to the mechanisms of action/toxicity induced by therapeutic monoclonal antibodies, antibody related products and biosimilars.
Public purpose
Underlining problems related to cardiac dysfunction caused by antibody-based HER2 targeted therapies are very important issues related to women’s health. Innovative methods to detect potential cause(s) of cardiotoxicity are urgently needed to improve the quality of life for women with HER2-positive breast cancer. Recent data obtained from several large clinical trials have demonstrated that cardiomyopathy induced by trastuzumab is irreversible in many patients. Therefore, it is important to develop sensitive and specific testing methods and biomarkers that could detect potential cardiotoxicity induced by trastuzumab therapy to provide important information to support effective post-marketing surveillance program.
Objective/specific aim
1. Clinical development of cMLC-1 as a novel predictive biomarker for trastuzumab-induced
cardiotoxicity.
2. Investigating the mechanistic interaction between anthracycline and trastuzumab and its clinical implications and developing biomarkers for trastuzumab-induced cardiotoxicity
Detailed Description
Clinical study at MGH/Harvard:
1. The grantee will first establish the assay to quantitatively measure cMLC-1 in human.
2. The grantee will investigate the value of cMLC-1 in humans as an early predictor of cardiotoxicity in a pilot study.
3. The archived plasma samples collected at multiple time points indicated below schematically will be tested for our biomarker cMLC-1 and for troponin I, an established marker for heart muscle damage as a control.
Study performed at FDA:
1) Investigate the functional connections between trastuzumab-induced cardiac toxicity and cMLC-1 in human primary cardiomyocytes.
- Effects of trastuzumab (10 mg/mL) and trastuzumab (10 mg/mL) plus pertuzumab (10 mg/mL) (T+P) on cardiomyocyte viability, mitochondria integrity, and contractility:
- Trastuzumab-mediated release of cMLC-1 and troponin 1 to the cell culture media:
- Trastuzumab-mediated signaling alterations in cardiomyocytes:
2) Investigate the mechanistic interaction between anthracycline and trastuzumab and monitoring cMLC1 release cell culture media and mouse serum.
SeeSection VIII. Other Information for award authorities and regulations.Grant: support mechanism providing money, property, or both to an eligible entity to carry out an approved project or activity.
New
The OER
Glossary and the SF424 (R&R) Application Guide provide details on
these application types.
The number of awards is contingent upon FDA appropriations and the submission of a sufficient number of meritorious applications. Award(s) will provide two (2) years of support
FDA/CDER intends to fund up to $140,000, for a one year period of performance in support of this grant program. FDA/CDER intends to fund up to $140,000 for fiscal year 2017 in support of this grant program.
It is anticipated that 1 award will be made, not to exceed $140,000 in total costs (direct plus indirect) over the course of the grant.
Application budgets need to reflect the actual needs of the proposed project and should not exceed the following in total costs (direct and indirect):
YR 01: $140,000.00
The scope of the proposed project should determine the project period. The maximum project period is (2) years.
HHS grants policies as described in the HHS Grants Policy Statement will apply to the applications submitted and awards made in response to this FOA.
Eligible Organizations
Higher Education Institutions
- Public/State Controlled Institutions of Higher Education
- Private Institutions of Higher Education
The following types of Higher Education Institutions are always encouraged to apply for FDA support as Public or Private Institutions of Higher Education:
o Hispanic-serving Institutions
o Historically Black Colleges and Universities (HBCUs)
o Tribally Controlled Colleges and Universities (TCCUs)
o Alaska Native and Native Hawaiian Serving Institutions
o Asian American Native American Pacific Islander Serving Institutions (AANAPISIs)
Nonprofits Other Than Institutions of Higher Education
- Nonprofits with 501(c)(3) IRS Status (Other than Institutions of Higher Education)
- Nonprofits without 501(c)(3) IRS Status (Other than Institutions of Higher Education)
For-Profit Organizations
- Small Businesses
- For-Profit Organizations (Other than Small Businesses)
Governments
- State Governments
- County Governments
- City or Township Governments
- Special District Governments
- Indian/Native American Tribal Governments (Federally Recognized)
- Indian/Native American Tribal Governments (Other than Federally Recognized)
- Eligible Agencies of the Federal Government
- U.S. Territory or Possession
Other
- Independent School Districts
- Public Housing Authorities/Indian Housing Authorities
- Native American Tribal Organizations (other than Federally recognized tribal governments)
- Faith-based or Community-based Organizations
- Regional Organizations
- Non-domestic (non-U.S.) Entities (Foreign Institutions)
Foreign Institutions
Non-domestic (non-U.S.) Entities (Foreign Institutions) are not eligible to apply.
Non-domestic (non-U.S.) components of U.S. Organizations are not eligible to apply.
Foreign components, as defined in the HHS Grants Policy Statement, are not allowed.
Applicant Organizations
Applicant organizations must complete and maintain the following registrations as described in the SF 424 (R&R) Application Guide to be eligible to apply for or receive an award. All registrations must be completed prior to the application being submitted. Registration can take 6 weeks or more, so applicants should begin the registration process as soon as possible. Failure to complete registrations in advance of a due date is not a valid reason for a late submission.
- Dun and Bradstreet Universal Numbering System (DUNS) - All registrations require that applicants be issued a DUNS number. After obtaining a DUNS number, applicants can begin both SAM and eRA Commons registrations. The same DUNS number must be used for all registrations, as well as on the grant application.
- System for Award Management (SAM) (formerly CCR) Applicants must complete and maintain an active registration, which requires renewal at least annually. The renewal process may require as much time as the initial registration. SAM registration includes the assignment of a Commercial and Government Entity (CAGE) Code for domestic organizations which have not already been assigned a CAGE Code.
- NATO Commercial and Government Entity (NCAGE) Code Foreign organizations must obtain an NCAGE code (in lieu of a CAGE code) in order to register in SAM.
- eRA Commons - Applicants must have an active DUNS number and SAM registration in order to complete the eRA Commons registration. Organizations can register with the eRA Commons as they are working through their SAM or Grants.gov registration. eRA Commons requires organizations to identify at least one Signing Official (SO) and at least one Program Director/Principal Investigator (PD/PI) account in order to submit an application.
- Grants.gov Applicants must have an active DUNS number and SAM registration in order to complete the Grants.gov registration.
Program Directors/Principal Investigators (PD(s)/PI(s))
All PD(s)/PI(s) must have an eRA Commons account. PD(s)/PI(s) should work with their organizational officials to either create a new account or to affiliate their existing account with the applicant organization in eRA Commons. If the PD/PI is also the organizational Signing Official, they must have two distinct eRA Commons accounts, one for each role. Obtaining an eRA Commons account can take up to 2 weeks.
Any individual(s) with the skills, knowledge, and resources necessary to carry out the proposed research as the Program Director(s)/Principal Investigator(s) (PD(s)/PI(s)) is invited to work with his/her organization to develop an application for support. Individuals from underrepresented racial and ethnic groups as well as individuals with disabilities are always encouraged to apply for FDA support.
For institutions/organizations proposing multiple PDs/PIs, visit the Multiple Program Director/Principal Investigator Policy and submission details in the Senior/Key Person Profile (Expanded) Component of the SF424 (R&R) Application Guide.
This FOA does not require cost sharing as defined in the HHS Grants Policy Statement.
Applicant organizations may submit more than one application, provided that each application is scientifically distinct.
The FDA will not accept duplicate or highly overlapping applications under review at the same time. This means that the FDA will not accept:
- A new (A0) application that is submitted before issuance of the summary statement from the review of an overlapping new (A0) or resubmission (A1) application.
- A resubmission (A1) application that is submitted before issuance of the summary statement from the review of the previous new (A0) application.
Buttons to access the online ASSIST system or to download application forms are available in Part 1 of this FOA. See your administrative office for instructions if you plan to use an institutional system-to-system solution.
It is critical that applicants follow the instructions in the Research Instructions for the SF424 (R&R) Application Guide, including Supplemental Grant Application Instructions except where instructed in this funding opportunity announcement to do otherwise. Conformance to the requirements in the Application Guide is required and strictly enforced. Applications that are out of compliance with these instructions may be delayed or not accepted for review.
For information on Application Submission and Receipt, visit Frequently Asked Questions Application Guide, Electronic Submission of Grant Applications.
All page limitations described in the SF424 Application Guide and the Table of Page Limits must be followed, with the following exceptions or additional requirements:
- For this specific FOA, the Research Strategy section is limited to 30 pages.
The following section supplements the instructions found in the SF424 (R&R) Application Guide and should be used for preparing an application to this FOA.
All instructions in the SF424 (R&R) Application Guide must be followed.
All instructions in the SF424 (R&R) Application Guide must be followed.
All instructions in the SF424 (R&R) Application Guide must be followed.
All instructions in the SF424 (R&R) Application Guide must be followed.
All instructions in the SF424 (R&R) Application Guide must be followed with the following additional instructions:
- Applications requesting multiple years of support must complete and submit a separate detailed budget breakdown and narrative justification for each year of financial support requested.
- If an applicant is requesting indirect costs as part of their budget, a copy of the most recent Federal indirect cost rate or F&A agreement must be provided as part of the application submission. This agreement should be attached to the RESEARCH & RELATED Other Project Information Component as line #12 'Other Attachments'.
- If the applicant organization has never established an indirect cost rate and/or does not have a negotiated Federal indirect cost rate agreement, a de minimis indirect cost rate of 10 percent (10%) of modified total direct costs (MTDC) will be allowed. MTDC means all direct salaries and wages, applicable fringe benefits, materials and supplies, services, travel, and subaward and subcontracts up to the first $25,000 of each subaward or subcontract. MTDC excludes equipment, capital expenditures, charges for patient care, rental costs, tuition remission, scholarships and fellowships, participant support costs and the portion of each subaward and subcontract in excess of $25,000.
All instructions in the SF424 (R&R) Application Guide must be followed.
All instructions in the SF424 (R&R) Application Guide must be followed.
All instructions in the SF424 (R&R) Application Guide must be followed.
Resource Sharing Plan: Individuals are required to comply with the instructions for the Resource Sharing Plans as provided in the SF424 (R&R) Application Guide, with the following modification:
- All applications, regardless of the amount of direct costs requested for any one year, should address a Data Sharing Plan.
Appendix: Do not use the Appendix to circumvent page limits. Follow all instructions for the Appendix as described in the SF424 (R&R) Application Guide.
When conducting clinical research, follow all instructions for completing PHS Inclusion Enrollment Report as described in the SF424 (R&R) Application Guide.
All instructions in the SF424 (R&R) Application Guide must be followed.
Not allowed
See Part 1. Section III.1 for information regarding the requirement for obtaining a unique entity identifier and for completing and maintaining active registrations in System for Award Management (SAM), NATO Commercial and Government Entity (NCAGE) Code (if applicable), eRA Commons, and Grants.gov
Part I. Overview Information contains information about Key Dates and times. Applicants are encouraged to submit applications before the due date to ensure they have time to make any application corrections that might be necessary for successful submission.
Organizations must submit applications to Grants.gov (the online portal to find and apply for grants across all Federal agencies). Applicants must then complete the submission process by tracking the status of the application in the eRA Commons, FDA's electronic system for grants administration. eRA Commons and Grants.gov systems check the application against many of the application instructions upon submission. Errors must be corrected and a changed/corrected application must be submitted to Grants.gov on or before the application due date and time. If a Changed/Corrected application is submitted after the deadline, the application will be considered late. Late applications will not be accepted for this FOA.
Applicants are responsible for viewing their application before the due date in the eRA Commons to ensure accurate and successful submission.
Information on the submission process and a definition of on-time submission are provided in the SF424 (R&R) Application Guide.
This initiative is not subject to intergovernmental review.
All FDA awards are subject to the terms and conditions, cost principles, and other considerations described in the HHS Grants Policy Statement.
Pre-award costs are allowable only as described in the HHS Grants Policy Statement.
Additional funding restrictions may be part of the Notice of Award.
Applications must be submitted electronically following the instructions described in the SF424 (R&R) Application Guide. Paper applications will not be accepted.
Applicants must complete all required registrations before the application due date. Section III. Eligibility Information contains information about registration.
For assistance with your electronic application or for more information on the electronic submission process, visit Applying Electronically. For assistance with application submission, contact the Application Submission Contacts in Section VII.
Important reminders:
All PD(s)/PI(s) must include their eRA Commons ID in the Credential field of the Senior/Key Person Profile Component of the SF424(R&R) Application Package. Failure to register in the Commons and to include a valid PD/PI Commons ID in the credential field will prevent the successful submission of an electronic application to FDA. See Section III of this FOA for information on registration requirements.
The applicant organization must ensure that the DUNS number it provides on the application is the same number used in the organization’s profile in the eRA Commons and for the System for Award Management. Additional information may be found in the SF424 (R&R) Application Guide.
See more tips for avoiding common errors.
Upon receipt, applications will be evaluated for completeness and compliance with application instructions by the assigned Grants Management Specialist and responsiveness by components of participating organizations, FDA. Applications that are incomplete, non-compliant and/or nonresponsive will not be reviewed.
Applicants are required to follow the instructions for post-submission materials, as described in NOT-OD-13-030.
Only the review criteria described below will be considered in the review process.
Reviewers will consider each of the review criteria below in the determination of scientific merit.
Does the project address an important problem or a critical barrier to progress in the field? Is there a strong scientific premise for the project? If the aims of the project are achieved, how will scientific knowledge, technical capability, and/or clinical practice be improved? How will successful completion of the aims change the concepts, methods, technologies, treatments, services, or preventative interventions that drive this field?
Are the PD(s)/PI(s), collaborators, and other researchers well suited to the project? If Early Stage Investigators or New Investigators, or in the early stages of independent careers, do they have appropriate experience and training? If established, have they demonstrated an ongoing record of accomplishments that have advanced their field(s)? If the project is collaborative or multi-PD/PI, do the investigators have complementary and integrated expertise; are their leadership approach, governance and organizational structure appropriate for the project?
Does the application challenge and seek to shift current research or clinical practice paradigms by utilizing novel theoretical concepts, approaches or methodologies, instrumentation, or interventions? Are the concepts, approaches or methodologies, instrumentation, or interventions novel to one field of research or novel in a broad sense? Is a refinement, improvement, or new application of theoretical concepts, approaches or methodologies, instrumentation, or interventions proposed?
Are the overall strategy, methodology, and analyses well-reasoned and appropriate to accomplish the specific aims of the project? Have the investigators presented strategies to ensure a robust and unbiased approach, as appropriate for the work proposed? Are potential problems, alternative strategies, and benchmarks for success presented? If the project is in the early stages of development, will the strategy establish feasibility and will particularly risky aspects be managed? Have the investigators presented adequate plans to address relevant biological variables, such as sex, for studies in vertebrate animals or human subjects?
If the project involves human subjects and/or FDA-defined clinical research, are the plans to address 1) the protection of human subjects from research risks, and 2) inclusion (or exclusion) of individuals on the basis of sex/gender, race, and ethnicity, as well as the inclusion or exclusion of children, justified in terms of the scientific goals and research strategy proposed?
Will the scientific environment in which the work will be done contribute to the probability of success? Are the institutional support, equipment and other physical resources available to the investigators adequate for the project proposed? Will the project benefit from unique features of the scientific environment, subject populations, or collaborative arrangements?
As applicable for the project proposed, reviewers will evaluate the following additional items, but will not give separate scores for these items and should not consider them in providing an overall score.
For research that involves human subjects but does not involve one of the six categories of research that are exempt under 45 CFR Part 46, the committee will evaluate the justification for involvement of human subjects and the proposed protections from research risk relating to their participation according to the following five review criteria: 1) risk to subjects, 2) adequacy of protection against risks, 3) potential benefits to the subjects and others, 4) importance of the knowledge to be gained, and 5) data and safety monitoring for clinical trials.
For research that involves human subjects and meets the criteria for one or more of the six categories of research that are exempt under 45 CFR Part 46, the committee will evaluate: 1) the justification for the exemption, 2) human subjects involvement and characteristics, and 3) sources of materials. For additional information on review of the Human Subjects section, please refer to the Guidelines for the Review of Human Subjects.
When the proposed project involves human subjects and/or FDA-defined clinical research, the committee will evaluate the proposed plans for the inclusion (or exclusion) of individuals on the basis of sex/gender, race, and ethnicity, as well as the inclusion (or exclusion) of children to determine if it is justified in terms of the scientific goals and research strategy proposed. For additional information on review of the Inclusion section, please refer to the Guidelines for the Review of Inclusion in Clinical Research.
The committee will evaluate the involvement of live vertebrate animals as part of the scientific assessment according to the following criteria: (1) description of proposed procedures involving animals, including species, strains, ages, sex, and total number to be used; (2) justifications for the use of animals versus alternative models and for the appropriateness of the species proposed; (3) interventions to minimize discomfort, distress, pain and injury; and (4) justification for euthanasia method if NOT consistent with the AVMA Guidelines for the Euthanasia of Animals. Reviewers will assess the use of chimpanzees as they would any other application proposing the use of vertebrate animals. For additional information on review of the Vertebrate Animals section, please refer to the Worksheet for Review of the Vertebrate Animal Section.
Reviewers will assess whether materials or procedures proposed are potentially hazardous to research personnel and/or the environment, and if needed, determine whether adequate protection is proposed.
Not Applicable.
Not Applicable.
Not Applicable
No.
Reviewers will assess the information provided in this section of the application, including 1) the Select Agent(s) to be used in the proposed research, 2) the registration status of all entities where Select Agent(s) will be used, 3) the procedures that will be used to monitor possession use and transfer of Select Agent(s), and 4) plans for appropriate biosafety, biocontainment, and security of the Select Agent(s).
Reviewers will comment on whether the following Resource Sharing Plans, or the rationale for not sharing the following types of resources, are reasonable: (1) Data Sharing Plan; (2) Sharing Model Organisms; and (3) Genomic Data Sharing Plan (GDS).
For projects involving key biological and/or chemical resources, reviewers will comment on the brief plans proposed for identifying and ensuring the validity of those resources.
Reviewers will consider whether the budget and the requested period of support are fully justified and reasonable in relation to the proposed research.
Applications will be evaluated for scientific and technical merit by an Objective Review Committee using the stated review criteria.
As part of the objective review, all applications:
- Will receive a written critique.
Appeals of objective review will not be accepted for applications submitted in response to this FOA.
Applications will compete for available funds with all other recommended applications submitted in response to this FOA. The following will be considered in making funding decisions:
- Scientific and technical merit of the proposed project as determined by objective review.
- Availability of funds.
- Relevance of the proposed project to program priorities.
After the Ad Hoc Review of the application is completed, the PD/PI will be able to access his or her Summary Statement (written critique) via E-mail from the Grants Management Specialist. The decision not to award a grant, or to award a grant at a particular funding level, is discretionary and is not subject to appeal to any FDA or HHS official or board. Information regarding the disposition of applications is available in the HHS Grants Policy Statement.
A formal notification in the form of a Notice of Award (NoA) will be provided to the applicant organization for successful applications. The NoA signed by the grants management officer is the authorizing document and will be sent via email to the grantee’s business official.
Awardees must comply with any funding restrictions described in Section IV.5. Funding Restrictions. Selection of an application for award is not an authorization to begin performance. Any costs incurred before receipt of the NoA are at the recipient's risk. These costs may be reimbursed only to the extent considered allowable pre-award costs.
Any application awarded in response to this FOA will be subject to terms and conditions found in the HHS Grants Policy Statement.
All FDA grant and cooperative agreement awards include the HHS Grants Policy Statement as part of the NoA.
Recipients of federal financial assistance (FFA) from HHS must administer their programs in compliance with federal civil rights law. This means that recipients of HHS funds must ensure equal access to their programs without regard to a person’s race, color, national origin, disability, age and, in some circumstances, sex and religion. This includes ensuring your programs are accessible to persons with limited English proficiency. HHS recognizes that research projects are often limited in scope for many reasons that are nondiscriminatory, such as the principal investigator’s scientific interest, funding limitations, recruitment requirements, and other considerations. Thus, criteria in research protocols that target or exclude certain populations are warranted where nondiscriminatory justifications establish that such criteria are appropriate with respect to the health or safety of the subjects, the scientific study design, or the purpose of the research.
In accordance with the statutory provisions contained in Section 872 of the Duncan Hunter National Defense Authorization Act of Fiscal Year 2009 (Public Law 110-417), FDA awards will be subject to the Federal Awardee Performance and Integrity Information System (FAPIIS) requirements. FAPIIS requires Federal award making officials to review and consider information about an applicant in the designated integrity and performance system (currently FAPIIS) prior to making an award. An applicant, at its option, may review information in the designated integrity and performance systems accessible through FAPIIS and comment on any information about itself that a Federal agency previously entered and is currently in FAPIIS. The Federal awarding agency will consider any comments by the applicant, in addition to other information in FAPIIS, in making a judgment about the applicant s integrity, business ethics, and record of performance under Federal awards when completing the review of risk posed by applicants as described in 45 CFR Part 75.205 Federal awarding agency review of risk posed by applicants. This provision will apply to all FDA grants and cooperative agreements.
HHS provides general guidance to recipients of FFA on meeting their legal obligation to take reasonable steps to provide meaningful access to their programs by persons with limited English proficiency. Please see http://www.hhs.gov/ocr/civilrights/resources/laws/revisedlep.html. The HHS Office for Civil Rights also provides guidance on complying with civil rights laws enforced by HHS. Please see http://www.hhs.gov/ocr/civilrights/understanding/section1557/index.html; and http://www.hhs.gov/ocr/civilrights/understanding/index.html. Recipients of FFA also have specific legal obligations for serving qualified individuals with disabilities. Please see http://www.hhs.gov/ocr/civilrights/understanding/disability/index.html. Please contact the HHS Office for Civil Rights for more information about obligations and prohibitions under federal civil rights laws at http://www.hhs.gov/ocr/office/about/rgn-hqaddresses.html or call 1-800-368-1019 or TDD 1-800-537-7697. Also note it is an HHS Departmental goal to ensure access to quality, culturally competent care, including long-term services and supports, for vulnerable populations. For further guidance on providing culturally and linguistically appropriate services, recipients should review the National Standards for Culturally and Linguistically Appropriate Services in Health and Health Care at http://minorityhealth.hhs.gov/omh/browse.aspx?lvl=2&lvlid=53.
FDA considers the sharing of research resources developed through FDA-sponsored research an important means to enhance the value and further the advancement of research. When research resources have been developed with FDA funds and the associated research findings published, those findings must be made readily available to the scientific community.
Upon acceptance for publication, scientific researchers must submit the author’s final manuscript of the peer-reviewed scientific publication resulting from research supported in whole or in part with FDA funds to the NIH National Library of Medicine's (NLM) PubMed Central (PMC). FDA defines the author's final manuscript as the final version accepted for journal publication, which includes all modifications from the publishing peer review process. The PMC archive is the designated repository for these manuscripts for use by the public, health care providers, educators, scientists, and FDA. Please see the FDA Public Access Policy.
Additional terms and conditions regarding FDA regulatory and FDA CENTER NAME programmatic requirements may be part of the Notice of Award.
Cooperative Agreement Terms and Conditions of Award
Clinical investigators at the clinical sites will conduct proposed clinical research and communicate with FDA research team about the progress/results, including all the raw data and analyzed data obtained during the project period.
Dr. Wen Jin Wu, the investigator at FDA, will collaborate with the clinical investigator and discuss the proposed study and interpret and analyze data obtained from the clinical study.
STANDARD TERMS AND CONDITIONS:
Financial Reporting:
A. Cash Transaction Reports
The Federal Financial Report (FFR) has a dedicated section to report Federal cash receipts and disbursements. For recipients this information must be submitted quarterly directly to the Payment Management System (PMS) using the web-based tool. Quarterly reports are due 30 days following the end of each calendar quarter. The reporting period for this report continues to be based on the calendar quarter. Questions concerning the requirements for this quarterly financial report should be directed to the PMS.
B. Financial Expenditure Reports
A required Federal Financial Report (FFR) must be submitted annually. FDA now requires all annual financial expenditure reports to be submitted electronically using the Federal Financial Report (FFR) system located in the eRA Commons. This includes all initial FFRs being prepared for submission and any revised FSR/FFRs being submitted or re-submitted to FDA. Paper expenditure/FFR reports will not accepted.
Annual FFRs must be submitted for each budget period no later than 90 days after the end of the calendar quarter in which the budget period ended. The reporting period for an annual FFR will be that of the budget period for the particular grant; however, the actual submission date is based on the calendar quarter. Failure to submit timely reports may affect future funding.
Performance Progress Reporting:
1. Annual progress reports are required. The Annual Progress Report will be due as part of the Research Performance Progress Report (RPPR).
2. Grants with Multiple Years: When multiple years are involved, awardees will be required to submit the Research Performance Progress Report (RPPR).
Information regarding submitting the RPPR is available at https://era.nih.gov/erahelp/commons/default.htm#cshid=1020
FAILURE TO COMPLY WITH THE ABOVE STATED TERMS AND CONDITIONS COULD RESULT IN THE SUSPENSION OR TERMINATION OF THIS COOPERATIVE AGREEMENT.
All formal correspondence/reports regarding the grant should be signed by an authorized institutional official and the Principal Investigator and should be sent to the attention of the grants management specialist, unless otherwise directed.
When multiple years are involved, awardees will be required to submit the Research Performance Progress Report (RPPR) annually and financial statements as required in the Notice of Award.
A final progress report, invention statement, and the expenditure data portion of the Federal Financial Report are required for closeout of an award, as described in the HHS Grants Policy Statement.
The Federal Funding Accountability and Transparency Act of 2006 (Transparency Act), includes a requirement for awardees of Federal grants to report information about first-tier subawards and executive compensation under Federal assistance awards issued in FY2011 or later. All awardees of applicable FDA grants and cooperative agreements are required to report to the Federal Subaward Reporting System (FSRS) available at www.fsrs.gov on all subawards over $25,000.
In accordance with the regulatory requirements provided at 45 CFR 75.113 and Appendix XII to 45 CFR Part 75, recipients that have currently active Federal grants, cooperative agreements, and procurement contracts from all Federal awarding agencies with a cumulative total value greater than $10,000,000 for any period of time during the period of performance of a Federal award, must report and maintain the currency of information reported in the System for Award Management (SAM) about civil, criminal, and administrative proceedings in connection with the award or performance of a Federal award that reached final disposition within the most recent five-year period. The recipient must also make semiannual disclosures regarding such proceedings. Proceedings information will be made publicly available in the designated integrity and performance system (currently FAPIIS). This is a statutory requirement under section 872 of Public Law 110-417, as amended (41 U.S.C. 2313). As required by section 3010 of Public Law 111-212, all information posted in the designated integrity and performance system on or after April 15, 2011, except past performance reviews required for Federal procurement contracts, will be publicly available. Full reporting requirements and procedures are found in Appendix XII to 45 CFR Part 75 Award Term and Conditions for Recipient Integrity and Performance Matters.
We encourage inquiries concerning this funding opportunity
and welcome the opportunity to answer questions from potential applicants.
eRA Service Desk (Questions regarding ASSIST, eRA Commons
registration, submitting and tracking an application, documenting system
problems that threaten submission by the due date, post submission issues)
Finding Help Online: http://grants.nih.gov/support/ (preferred method of contact)
Telephone: 301-402-7469 or 866-504-9552 (Toll Free)
Grants.gov
Customer Support (Questions
regarding Grants.gov registration and submission, downloading forms and
application packages)
Contact Center Telephone: 800-518-4726
Web ticketing system: https://grants-portal.psc.gov/ContactUs.aspx
Email: [email protected]
Wen Jin Wu, M.D., Ph.D.
Food and Drug Administration
Phone: 240-402-6715
Shashi Malhotra
Food and Drug Administration
Telephone: 240-402-7592
Email: [email protected]
Shashi Malhotra
Food and Drug Administration
Telephone: 240-402-7592
Email: [email protected]
All awards are subject to the terms and conditions, cost principles, and other considerations described in the HHS Grants Policy Statement.
Awards are made under the authorization of Sections 301 of the Public Health Service Act as amended (42 USC 241) and under Federal Regulations 42 CFR Part 52 and 45 CFR Part 75.
and Human Services (HHS)